RAxML - Randomized Axelerated Maximum Likelihood
New RAxML citation
When using RAxML please cite the following paper: A. Stamatakis: "RAxML Version 8: A tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies". In Bioinformatics, 2014, open access.
Latest code download
Get the most up-to-date RAxML version from github.
Documentation
- new RAxML version 8.0.0 manual PDF
- copy of the old v704 manual: PDF
- For a basic step by step tutorial using some more recent features see RAxML step-by-step tutorial
- For a basic step by step tutorial by Pavlos Pavlidis on how to install and run RAxML on a Linux cluster see RAxML on cluster step-by-step tutorial
- For a video explaining the evolutionary placement algorithm for short reads see Alexis talking about evolutionary placement of short reads
- Video of Alexis talking about evolutionary placement of short reads at the Joint Genome Institute
- Some nice slides by Wayne Pfeiffer (SDSC) on the hybrid MPI/Pthreads version of RAxML and the hybrid MPI/OpenMP version of MrBayes
- some useful slides by Nick Pattengale explaining the bootstrap convergence criteria implemented in RAxML
- Antonis Rokas has written a nice chapter about hands-on phylogeny reconstruction that uses RAxML as example program
- A tutorial on how to install and run RAxML on a MAC in less than a minute by Dave Carmean
User Support
Please send all your questions and feature request to the RAxML google group. Before posting, keep in mind that a google group actually has a search function! Emails to Exelixis lab members regarding RAxML will not be answered. Messages posted via github will also not be answered.
RAxML memory requirements
Since datasets are getting larger here is a formula to estimate RAxML memory requirements: Given an alignment of n taxa and m distinct patterns the memory consumption is approximately:
- MEM(AA+GAMMA) = (n-2) * m * (80 * 8) bytes
- MEM(AA+CAT) = (n-2) * m * (20 * 8) bytes
- MEM(DNA+GAMMA) = (n-2) * m * (16 * 8) bytes
- MEM(DNA+CAT) = (n-2) * m * (4 * 8) bytes
You may also use th on-line calculator below:
Required size:
Web-Servers for evolutionary placement of short reads
Web-Servers for phylogenetic placement of short sequence reads (including alignment and visualization tools):
- advanced Swiss Server with pre-computed trees, please cite this paper when using it
- basic German Server without pre-computed trees, please cite this paper when using it
Web-Servers for tree building
co-maintaned by Exelixis Lab:
- Vital IT unit of the Swiss Institute of Bioinformatics
- CIPRES portal at San Diego Supercomputer Center
-
New beta-version of
the CIPRES portal that provides a full workbench.
I recently found this youtube video (in Spanish) with a nice tutorial on how to use the CIPRES portal:
not maintained by the Exelixis Lab:
- Bioportal in Norway (University of Oslo)
- Trex on-line Web-Server at Université du Québec à Montréal
Graphical User Interfaces (GUIs)
- Daniele Silvestro and Ingo Michalak at the Senckenberg Museum and Research Center have started developing a GUI for RAxML that runs under MACs, Windows, and Linux. The code for the GUI is available here. Please send suggestions and comments to Daniele Silvestro at senckenberg de
- Jacek Kominek from the University of Gdansk in Poland has developed this nice GUI here
Helper Scripts and Tools
Phylogenetic Binning tool
File Conversion scripts
- shell script by Andre Aberer for fasta to phylip conversion
- matlab programs by Lowie Li for fasta to phylip and phylip to fasta conversion
Wrapper Scripts
- raxml_launch_serially.sh: A simple shell script that launches one job after the other awaiting for completion of each job.
- raxml_nexusPartConvert.pl: A Perl script that parses a partitioned alignment in Nexus format with charsets and produces a partition guide file to be fed to RAxML with -q. Preliminary - works with DNA or AA, but not the two together yet, so not suitable for mixed-molecule data. Unless the output gets redirected to a file with ">", it will appear on screen.
- raxml_wrapper.pl: A Perl script that reads a raxml.config file with common run parameters and executes a directory of Phylip alignment files in batch, then outputs the results in another directory. See the documentation with "perldoc ./raxml_wrapper.pl".
- Given a best-known ML tree, generate a number of Bootstrap replicates and just re-estimate the branch lengths for that given fixed tree topology on each Bootstrap replicate.
- To invoke the script call it as follows: "perl bsBranchLengths.pl alignmentFileName treeFileName numberOfReplicates". The script assumes that the RAxML executable is located in the directory where you execute it. Otherwise, if RAxML is located in your Linux/Unix path just replace every occurence of "./raxmlHPC" by "raxmlHPC" in the script. The bootstrapped trees with branch lengths will be written into a file called "bsTrees".
- This script is intended for use with programs that infer divergence time estimates.
- Here is a little perl-script that will automatically determine the best-scoring AA substitution model on a fixed starting tree. Note that raxmlHPC must be in your $PATH for this to work.
- For unpartitioned datasets execute it like this: perl ProteinModelSelection.pl alignmentFile.phylip > outfile The outfile will then contain the best-scoring AA model to use with RAxML.
- For partitioned datasets execute it like this: perl ProteinModelSelection.pl alignmentFile.phylip partitionData.txt > outfile The outfile will then contain the best-scoring AA model for every partition.
Olaf Bininda-Emonds has written batchRAxML.pl. This nice script by my good colleague from Munich times Olaf Bininda-Emonds provides a wrapper around RAxML to easily analyze a set of data files according to a common set of the search criteria. Also organizes the RAxML output into a set of subdirectories.
Frank Kauff has written PYRAXML2. Frank Kauff at University of Kaiserslautern (formerly at Duke University) has written this cool script that reads NEXUS-style data files and prepares the necessary input files and command-line options for RAxML-VI-HPC. You can download the BETA-version here: PYRAXML2 It requires PYTHON and BIOPYTHON to be installed on your computer.
Old RAxML code versions
- RAxML v7.2.8 alpha release source code available here
- RAxML v7.2.7 (alpha) available for download here
- RAxML v7.2.6 available for download here and here is a windows executable
- RAxML v7.2.5 (alpha) available for download here and here is a windows executable
- RAxML v7.2.4(alpha) available for download here
- RAxML v7.2.3 (alpha) available for download here
- RAxML v7.2.2 available for download here and download windows executable
- RAxML v7.2.1 (alpha) available for download here windows executable here
- RAxML v7.2.0 (alpha) available for download here
- RAxML v7.1.0 (alpha) available for download here
- RAxML v7.0.4 available for download here
- RAxML v7.0.3 available for download here
- Windows executable. Graham Jones has provided a nice PDF on How to run RAxML under XP and Vista.
- Mac executable (iMAC)
- Mac executable (iMAC Pthreads-version)
- Mac executable (PowerMac G5)
- Mac executable (PowerMac G5 Pthreads-version)
- RAxML-VI-HPC (version 2.2.3)and a comprehensive Manual (v2.2.3)
- RAxML-VI-HPC (version 2.0.2) and a comprehensive Manual (v2.0)
- RAxML-VI-HPC (version 1.0) and a comprehensive Manual (v1.0)
- RAxML-VI: Sequential program with significantly accelerated hill-climbing search algorithm for huge alignment data.
- RAxML-III: Sequential program, includes more models of nucleotide substitution than RAxML-II.
- RAxML-II: Sequential, Parallel, and Distributed implementation of RAxML with less model functionality.
On-line material for some old RAxML papers
Material (alignments) for 2008 Systematic Biology paper on the rapid bootstrap algorithm- test datasets available here
Material (test datasets) for 2007 Supercomputing paper on parallelizing RAxML on the IBM BlueGene/L
- test datasets available here
- Click here for a table with the experimental raw data
Material for HPCC05 paper: “Parallel Divide-and-Conquer Phylogeny Reconstruction by Maximum Likelihood”
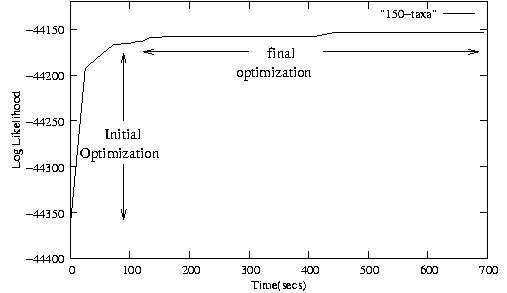
- Initial and final optimization phase of RAxML for an alignment with 150 sequences
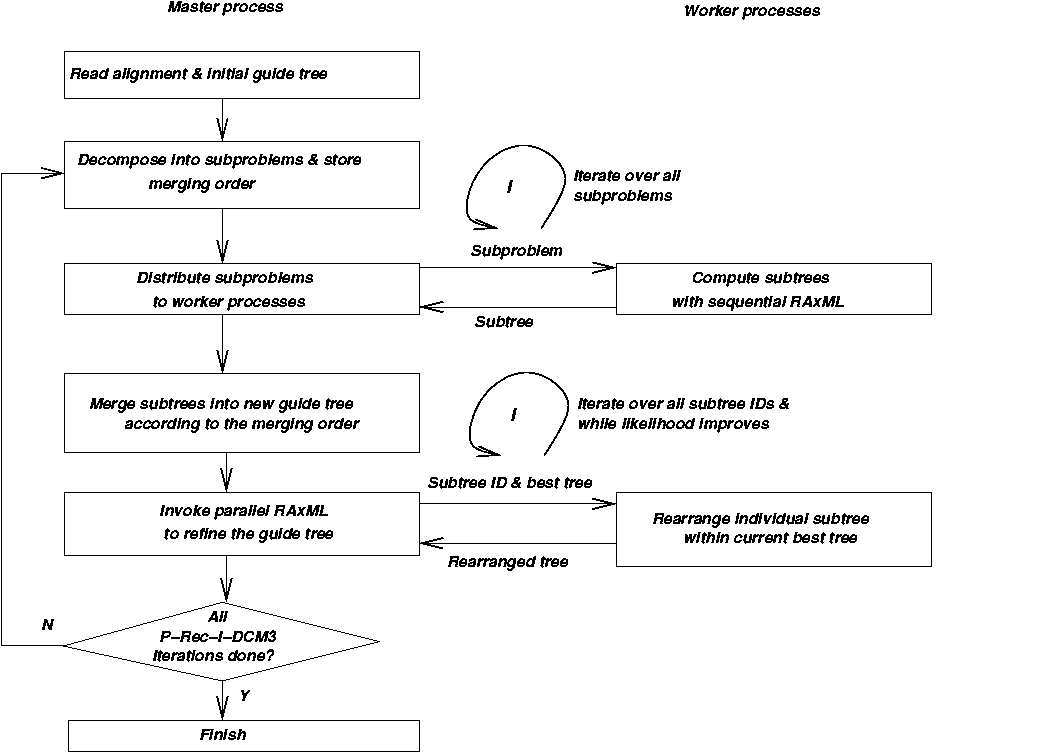
- Program Flow of P-Rec-I-DCM-3(RAxML)
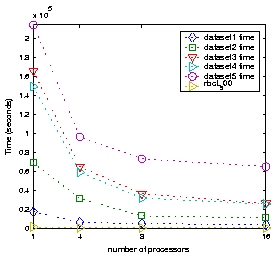
- Speedup value: Time to complete one iteration of P-Rec-I-DCM3 for datasets 1-5 and 1 up to 16 processors
{kind=link}
{kind=link}
{kind=link}
- 1,000 taxa plot alignment Alignment of 1,000 sequences
from the ARB database containing Eucarya, Bacteria, Archaea by Harald
Meier, TU München

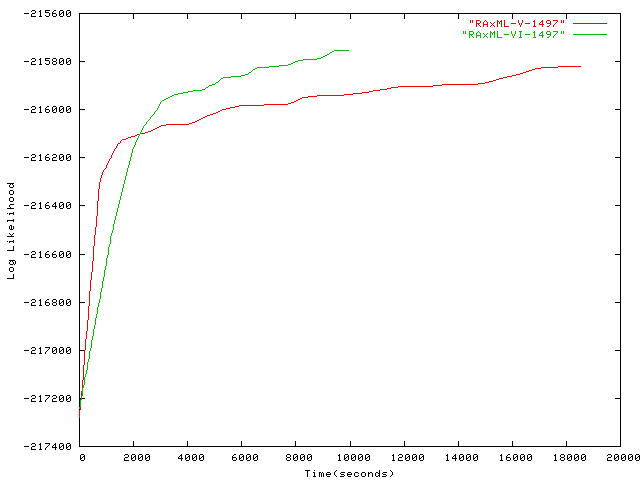
- 1,497 taxa plot Alignment of 1,497 Bacteria by Josh Wilcox, Pace Lab, University of Colorado at Boulder, for more information on this alignment please contact the Pace Lab
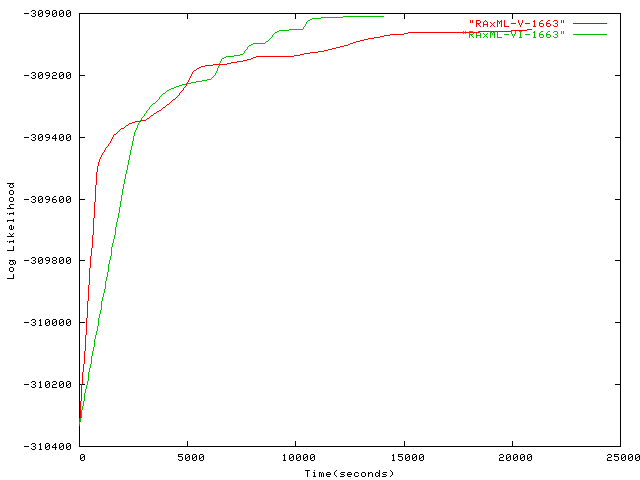
- 1,663 taxa plot alignment Alignment of 1,663 sequences from the ARB database containing Eucarya, Bacteria, Archaea by Harald Meier, TU München
- 1,728 taxa plot alignment Alignment of 1,728 Archaea by Chuck Robertson, Pace Lab, University of Colorado at Boulder
- 2,000 taxa plot alignment Ribosomal RNA sequences by Gutell Lab, University of Texas at Austin, for more information on this alignment please contact Robin Gutell
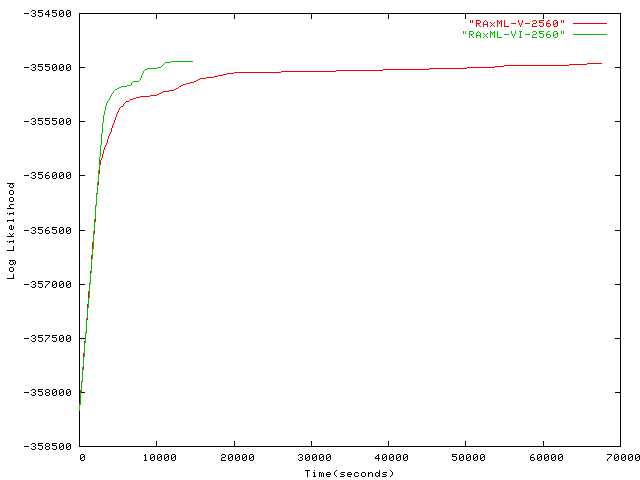
- 2,560 taxa plot alignment upon request via email Kallersjo, M., et al., Simultaneous parsimony jackknife analysis of 2538 rbcL DNA sequences reveals support for major clades of green plants, land plants, seed plants and flowering plants. Pl. Syst. Evol., 1998. 213: p. 259-287.
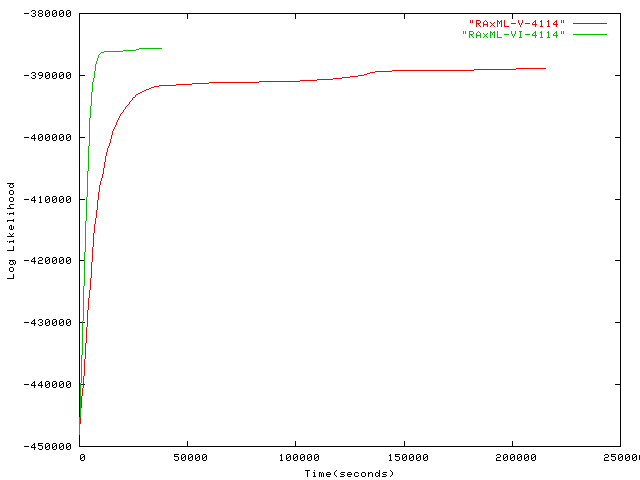
- 4,114 taxa plot alignment 16S ribosomal Actinobacteria RNA sequences, by Usman Roshan, New Jersey Institute of Technology
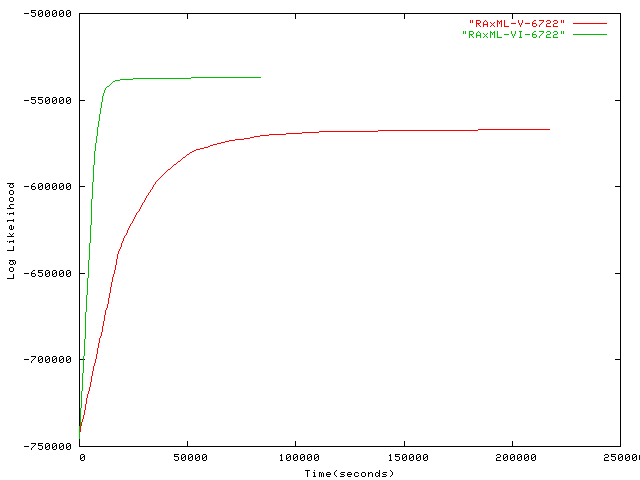
- 6,722 taxa plot alignment Ribosomal RNA sequences by Gutell Lab, University of Texas at Austin, for more information on this alignment please contact Robin Gutell
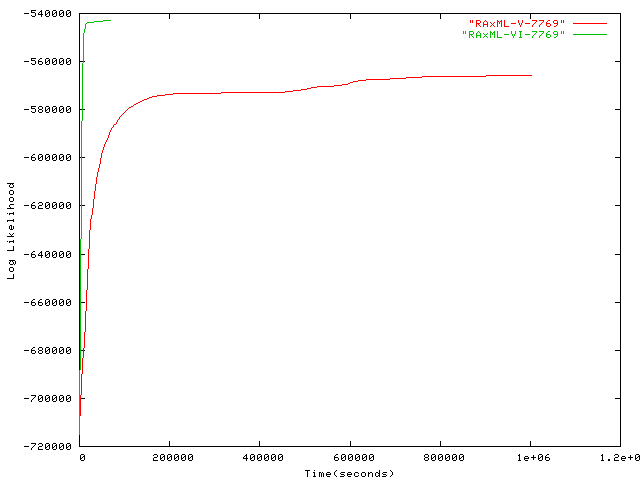
- 7,769 taxa plot alignment Ribosomal RNA sequences by Gutell Lab, University of Texas at Austin, for more information on this alignment please contact Robin Gutell
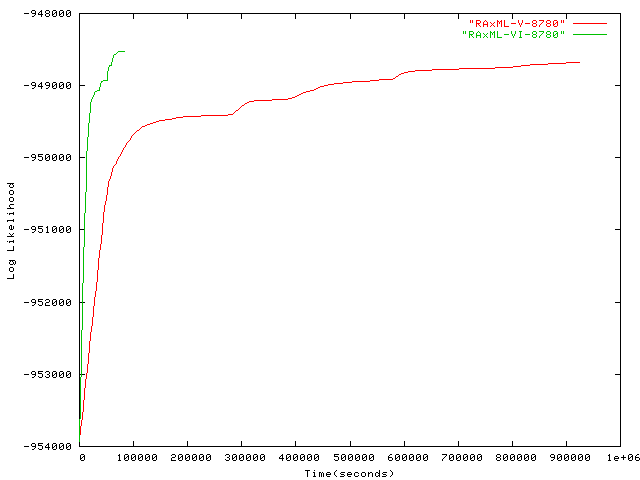
- 8,780 taxa plot alignment Alignment of 8,780 sequences from the ARB database containing Eucarya, Bacteria, Archaea. Original alignment by Harald Meier, TU München, modified by Usman Roshan, New Jersey Institute of Technology
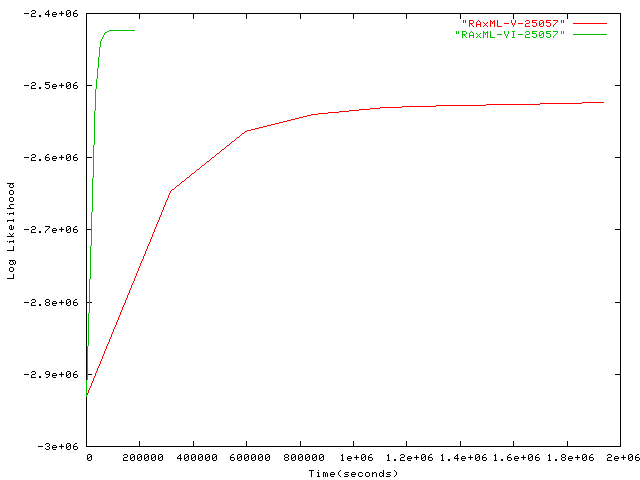
- 25,057 taxa plot alignment Alignment of 25,057 Protobacteria, by Usman Roshan, New Jersey Institute of Technology
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The old Alignment Benchmark set: includes some large real-world alignments and best-known trees for those alignments